 Close
Close

Head of Queen Square Brain Bank Professor Thomas Warner Clinical Associate Professor and Honorary Consultant Neuropathologist Dr Zane Jaunmuktane
Clinical Associate Professor and Honorary Consultant Neuropathologist Dr Karl Frontzek
“QSBB is an invaluable resource for clinicians and scientists worldwide. For 30 years we have built a unique collection of tissue focussing on neurodegenerative diseases. Our own research programme, and that of groups to whom we have supplied tissue for research, has resulted in a greater understanding of the underlying processes that lead to the death of specific brain cells, improvements in diagnostic precision, and opened new avenues for more effective treatments.”
Epigenetic age acceleration is associated with oligodendrocyte proportions in MSA and control brain tissue
Megha Murthy, Gemma Shireby, Yasuo Miki, Emmanuelle Viré, Tammaryn Lashley, Thomas T. Warner, Jonathan Mill, Conceição Bettencourt
"Neuropathology and Applied Neurobiology" December 2022
Abstract: Aims Epigenetic clocks are widely applied as surrogates for biological age in different tissues and/or diseases, including several neurodegenerative diseases. Despite white matter (WM) changes often being observed in neurodegenerative diseases, no study has investigated epigenetic ageing in white matter.
Results
Estimated DNA methylation (DNAm) ages showed strong correlations with chronological ages, even in WM (e.g., DNAmClockCortical, r = [0.80–0.97], p < 0.05). However, performances and DNAm age estimates differed between clocks and brain regions. DNAmClockMulti significantly underestimated ages in all cohorts except in the MSA prefrontal cortex mixed tissue, whereas DNAmClockCortical tended towards age overestimations. Pronounced age overestimations in the oligodendrocyte-enriched cohorts (e.g., oligodendrocyte-enriched nuclei, p = 6.1 × 10−5) suggested that this cell type ages faster. Indeed, significant positive correlations were observed between estimated oligodendrocyte proportions and DNAm age acceleration estimated by DNAmClockCortical (r > 0.31, p < 0.05), and similar trends were obtained with DNAmClockMulti. Although increased age acceleration was observed in MSA compared with controls, no significant differences were detected upon adjustment for possible confounders (e.g., cell-type proportions).
Conclusions
Our findings show that oligodendrocyte proportions positively influence epigenetic age acceleration across brain regions and highlight the need to further investigate this in ageing and neurodegeneration.
Fig. 1 Chronological and DNAm ages for DNAmClockMulti and DNAmClockCortical for the different brain regions of control and multiple system atrophy (MSA) samples in all cohorts. CT CRBL, control cerebellum (WM); CT FL, control frontal lobe (WM); CT OL, control occipital lobe (WM); CT PFC, control prefrontal cortex (WM + GM); WM + GM, mix of white and grey matter; MSA CRBL, multiple system atrophy (MSA) cerebellum (WM); MSA FL, MSA frontal lobe (WM); MSA OL, MSA occipital lobe (WM); MSA PFC, MSA prefrontal cortex (WM + GM); WM, white matter; the p-values were calculated using Wilcoxon signed rank exact test for paired samples.



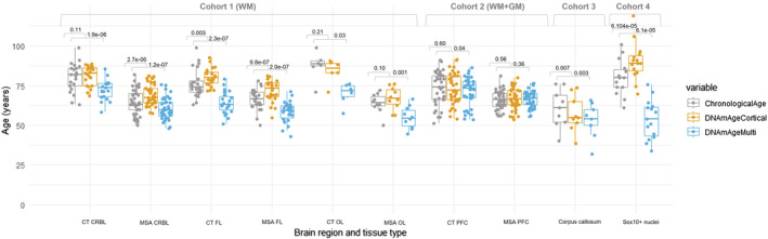
Fig. 2 Association between epigenetic age acceleration and oligodendrocyte proportions for DNAmClockCortical and DNAmClockMulti in the different brain regions. (A–D) Age acceleration residuals (y-axis) versus oligodendrocyte (SOX10+) proportions (x-axis) for DNAmClockCortical in the different brain regions; (E–H) age acceleration residuals (y-axis) versus oligodendrocyte (SOX10+) proportions (x-axis) for DNAmClockMulti in the different brain regions. Age acceleration residuals were obtained by regressing DNA methylation age against confounding factors, including chronological age; oligodendrocyte proportions were obtained using a DNA methylation-based cell-type deconvolution algorithm. The correlation coefficient and p-values shown were calculated using Pearson correlation. CC, control cerebellum (WM); CF, control frontal lobe (WM); CO, control occipital lobe (WM); CpC, corpus callosum; GM, grey matter; MC, multiple system atrophy (MSA) cerebellum (WM); MF, MSA frontal lobe (WM); MO, MSA occipital lobe (WM); PFC, prefrontal cortex (WM + GM); WM, white matter.
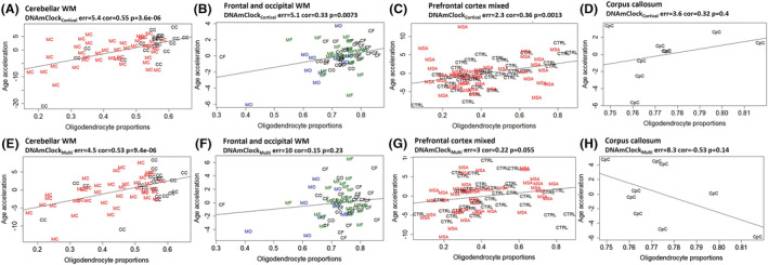
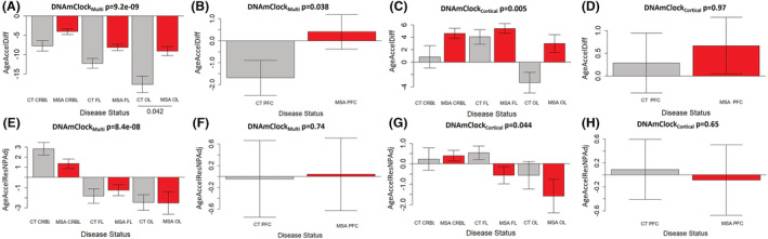
Fig. 3 Age acceleration estimates for DNAmClockCortical and DNAmClockMulti in the different brain regions. (A–D) Age acceleration difference for DNAmClockMulti (A—Cohort 1; B—Cohort 2) and DNAmClockCortical (C—Cohort 1; D—Cohort 2) in the different brain regions, (E–H) age acceleration residual after adjusting for chronological age and neuronal proportions for the DNAmClockMulti (E—Cohort 1; F—Cohort 2) and DNAmClockCortical (G—Cohort 1; H—Cohort 2) in the different brain regions. CT CRBL, control cerebellum (WM); CT FL, control frontal lobe (WM); CT OL, control occipital lobe (WM); CT PFC, control prefrontal cortex (WM + GM); GM, grey matter; MSA CRBL, multiple system atrophy (MSA) cerebellum (WM); MSA FL, MSA frontal lobe (WM); MSA OL, MSA occipital lobe (WM); MSA PFC, MSA prefrontal cortex (WM + GM); WM, white matter; the p-values for across group comparisons were calculated using the Kruskal–Wallis test, and p-values for pairwise analysis between MSA and controls for each brain region were calculated using the Wilcoxon's test with Benjamini–Hochberg correction for multiple testing
Clinical Diagnostic Accuracy of Parkinson’s Disease: Where Do We Stand?
Sasivimol Virameteekul, Tamas Revesz,Zane Jaunmuktane, Thomas T. Warner, Eduardo De Pablo-Fernandez
Abstract
Background and objectives: Clinical diagnostic accuracy of Parkinson’s disease (PD) remains suboptimal. Changes in disease concept may have improved clinical diagnostic accuracy in the past decade. However, current clinical diagnostic criteria have not been validated against neuropathological confirmation. Objectives: This study aims to provide up-to-date clinical diagnostic accuracy data and validate current clinical diagnostic criteria for PD against neuropathology.
Methods:A retrospective review of medical records of consecutive patients with parkinsonism from the Queen Square Brain Bank was performed between 2009 and 2019. Clinical diagnosis was documented at early (within 5 years of motor symptom onset) and final stages and categorized by movement disorder experts or regular clinicians. Movement Disorder Society Parkinson’s disease (MDS-PD) diagnostic criteria were retrospectively applied. Diagnostic accuracy parameters (sensitivity, specificity, positive/negative predictive value, and accuracy) were calculated using neuropathological diagnosis as the gold standard.
Results: A total of 267 patients (141 PD and 126 non- PD parkinsonism) were included. Clinical diagnostic accuracy was 97.2% for experts, 92.5% for the MDS clinically probable PD criteria, and 90.3% for clinicians.Similar figures were obtained when applied at an early stage (91.5%, 89.5%, and 84.2% diagnostic accuracy, respectively). MDS clinically established early PD criteria demonstrated very high specificity (98.4%) at early stages.
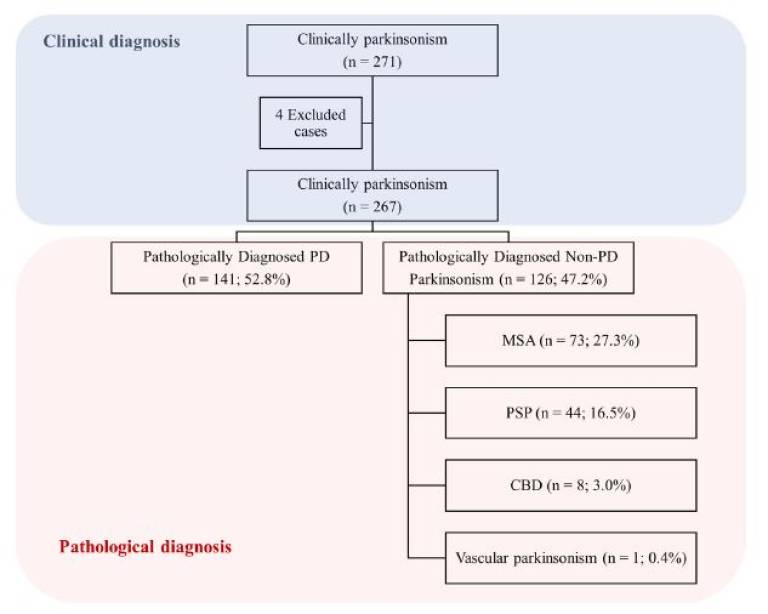 | Fig 1. Study design. PD, Parkinson’s disease; MSA, multiple system atrophy; PSP, progressive supranuclear palsy; CBD, corticobasal degeneration. |
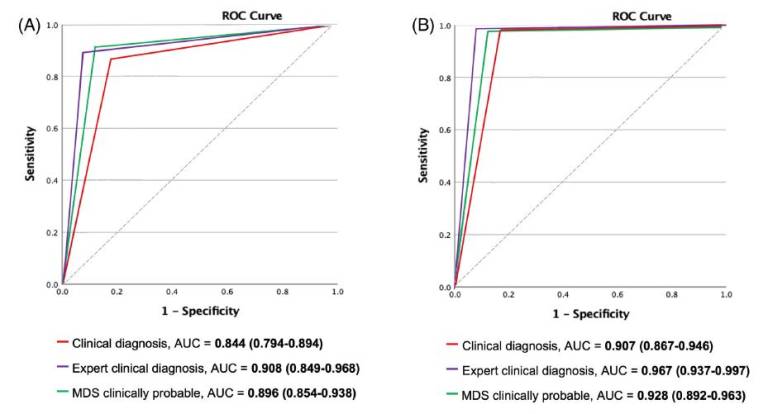 | Fig 2. ROC curves of each diagnostic category for Parkinson’s disease at (A) early and (B) final stages. AUC, area under the ROC curve; ROC, receiver operating characteristic. |
Brain DNA methylomic analysis of frontotemporal lobar degeneration reveals OTUD4 in shared dysregulated signatures across pathological subtypes
Katherine Fodder, Megha Murthy, Patrizia Rizzu, Christina E. Toomey, Rahat Hasan, Jack Humphrey, Towfque Raj, Katie Lunnon, Jonathan Mill, Peter Heutink, Tammaryn Lashley, Conceição Bettencourt
Acta Neuropathologica 07 May 2023
Abstract: Frontotemporal lobar degeneration (FTLD) is an umbrella term describing the neuropathology of a clinically, genetically and pathologically heterogeneous group of diseases, including frontotemporal dementia (FTD) and progressive supranuclear palsy (PSP). Among the major FTLD pathological subgroups, FTLD with TDP-43 positive inclusions (FTLD-TDP) and FTLD with tau-positive inclusions (FTLD-tau) are the most common, representing about 90% of the cases. Although alterations in DNA methylation have been consistently associated with neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease, little is known for FTLD and its heterogeneous subgroups and subtypes. The main goal of this study was to investigate DNA methylation variation in FTLD-TDP and FTLD-tau. We used frontal cortex genomewide DNA methylation profles from three FTLD cohorts (142 FTLD cases and 92 controls), generated using the Illumina 450K or EPIC microarrays. We performed epigenome-wide association studies (EWAS) for each cohort followed by metaanalysis to identify shared diferentially methylated loci across FTLD subgroups/subtypes. In addition, we used weighted gene correlation network analysis to identify co-methylation signatures associated with FTLD and other disease-related traits. Wherever possible, we also incorporated relevant gene/protein expression data. After accounting for a conservative Bonferroni multiple testing correction, the EWAS meta-analysis revealed two diferentially methylated loci in FTLD, one annotated to OTUD4 (5’UTR-shore) and the other to NFATC1 (gene body-island). Of these loci, OTUD4 showed consistent upregulation of mRNA and protein expression in FTLD. In addition, in the three independent co-methylation networks, OTUD4-containing modules were enriched for EWAS meta-analysis top loci and were strongly associated with the FTLD status. These co-methylation modules were enriched for genes implicated in the ubiquitin system, RNA/stress granule formation and glutamatergic synaptic signalling. Altogether, our fndings identified novel FTLD-associated loci, and support a role for DNA methylation as a mechanism involved in the dysregulation of biological processes relevant to FTLD, highlighting novel potential avenues for therapeutic development.
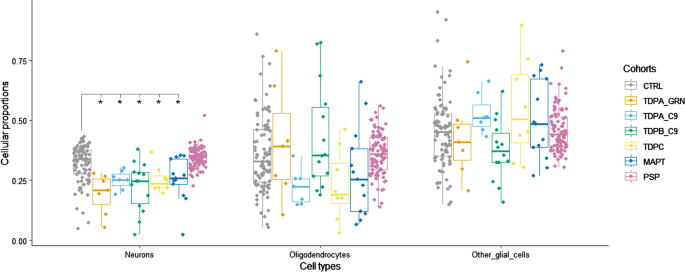 Fig 2. Brain cell-type proportion estimates derived from bulk DNA methylation data in frontal lobe of frontotemporal lobar degeneration (FTLD) and controls.*Indicates significant differences for each cell-type between FTLD subtypes and the corresponding controls; pairwise comparisons were performed using the Wilcoxon rank sum test, and adjusted p-values < 0.05 were considered significant. CTRL, controls; TDPA_GRN, FTLD with TDP-43 positive inclusions (FTLD-TDP) subtype A, carriers of GRN mutations; TDPA_C9, FTLD-TDP subtype A, carriers of C9orf72 repeat expansion; TDPB_C9, FTLD-TDP subtype B, carriers of C9orf72 repeat expansion; TDPC, FTLD-TDP subtype C, sporadic; MAPT, FTLD with tau-positive inclusions (FTLD-Tau), carriers of MAPT mutations; PSP, FTLD-Tau, sporadic progressive supranuclear palsy; Neurons, NeuN + ; Oligodendrocytes, SOX10 + ; other glial cells, NeuN−/SOX10− |
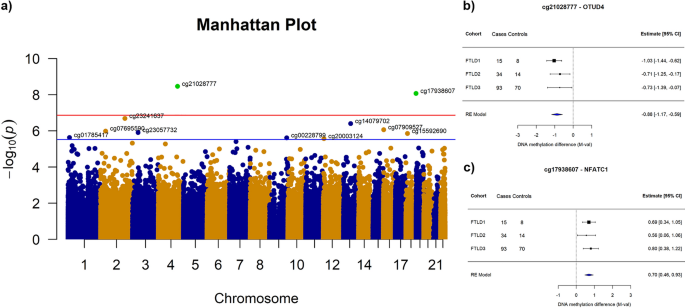
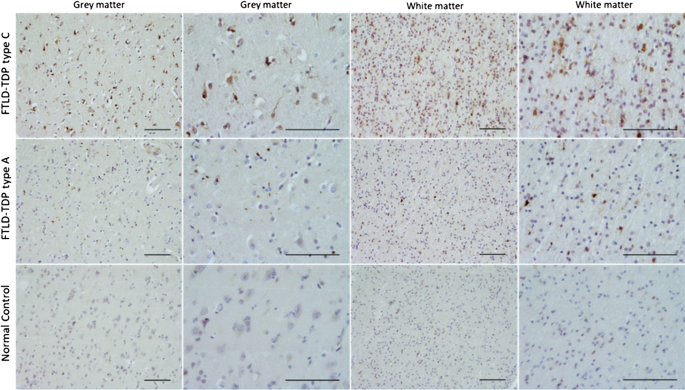
Fig 6. Immunoreactivity of OTUD4 in the frontal cortex of FTLD-TDP (N = 4 type A and N = 3 type C) and controls (N = 3). Immunohistochemical analysis was carried out in FFPE frontal cortex tissue from FTLD-TDP cases and controls overlapping with FTLD1, using a rabbit anti-OTUD4 antibody (Atlas Antibodies HPA036623, 1:200). Scale-bars represent 100 µm
Epigenetic Age Acceleration in Frontotemporal Lobar Degeneration: A Comprehensive Analysis in the Blood and Brain
Megha Murthy, Patrizia Rizzu, Peter Heutink, Jonathan Mill,Tammaryn Lashley,Conceição Bettencourt
Abstract: Frontotemporal lobar degeneration (FTLD) includes a heterogeneous group of disorders pathologically characterized by the degeneration of the frontal and temporal lobes. In addition to major genetic contributors of FTLD such as mutations in MAPT, GRN, and C9orf72, recent work has identified several epigenetic modifications including significant differential DNA methylation in DLX1, and OTUD4 loci. As aging remains one of the major risk factors for FTLD, we investigated the presence of accelerated epigenetic aging in FTLD compared to controls. We calculated epigenetic age in both peripheral blood and brain tissues of multiple FTLD subtypes using several DNA methylation clocks, i.e., DNAmClockMulti, DNAmClockHannum, DNAmClockCortical, GrimAge, and PhenoAge, and determined age acceleration and its association with different cellular proportions and clinical traits. Significant epigenetic age acceleration was observed in the peripheral blood of both frontotemporal dementia (FTD) and progressive supranuclear palsy (PSP) patients compared to controls with DNAmClockHannum, even after accounting for confounding factors. A similar trend was observed with both DNAmClockMulti and DNAmClockCortical in post-mortem frontal cortex tissue of PSP patients and in FTLD cases harboring GRN mutations. Our findings support that increased epigenetic age acceleration in the peripheral blood could be an indicator for PSP and to a smaller extent, FTD.
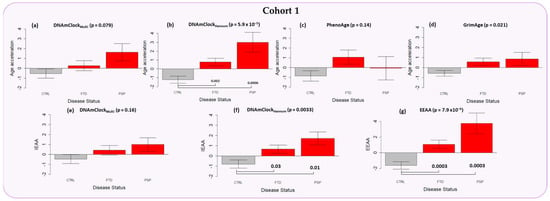
Figure 2. Epigenetic age acceleration in the peripheral blood samples of Cohort 1 (purple) constituting FTD (n = 117) and PSP cases (n = 44) as well as controls (n = 178) with the DNAmClockMulti, DNAmClockHannum, PhenoAge, and GrimAge clocks. (a–d) Epigenetic age acceleration (y-axis) in relation to disease status (x-axis) with the 4 clocks; (e,f) intrinsic epigenetic age acceleration (IEAA, y-axis) of DNAmClockMulti and DNAmClockHannum with respect to disease status (x-axis); and (g) extrinsic epigenetic age acceleration (EEAA, y-axis) with respect to disease status (x-axis). CTRL—control, FTD—frontotemporal dementia, PSP—progressive supranuclear palsy. Age acceleration residuals were obtained by regressing DNA methylation age against chronological age and adjusting for confounding factors such as cell type proportions. The bar plots depict the mean value and standard error (y-axis). p-values for across group comparisons were calculated using the Kruskal–Wallis test (p-values shown at the top of the plots (a–g)), and p-values for pairwise analysis between each disease group and controls were calculated using the Wilcoxon’s test with Benjamini–Hochberg correction for multiple testing (p-values shown at the bottom of the plots (a–g)). |
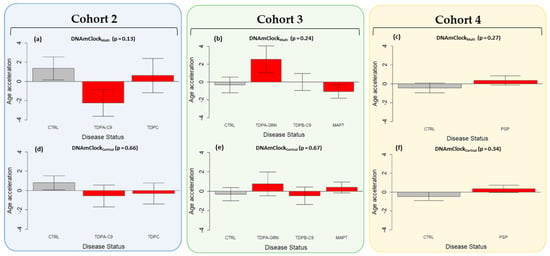
Figure 3. Epigenetic age acceleration in the post-mortem brain tissues using DNAmClockMulti and DNAmClockCortical. Cohort 2 (blue) constituted FTLD-TDP types A (C9orf72 mutation carriers, n = 7), and C (sporadic cases, n = 8), and controls (n = 8), Cohort 3 (green) constituted FTLD-TDP types B (C9orf72 mutation carriers n = 13), and A (GRN mutation carriers, n = 7), FTLD-Tau MAPT mutation carriers (n = 13) and controls (n = 14), and Cohort 4 (yellow) comprised PSP cases (n = 93) and controls (n = 71). (a–f) Epigenetic age acceleration (y-axis) in relation to disease status (x-axis) for the cohorts with DNAmClockMulti and DNAmClockCortical. CTRL—control, TDPA-C9—FTLD-TDPA (C9orf72 mutation carriers), TDPC—FTLD-TDPC (sporadic); TDPA-GRN—FTLD-TDPA (GRN mutation carriers), TDPB-C9—FTLD-TDPB (C9orf72 mutation carriers), MAPT—FTLD-Tau MAPT mutation carriers, PSP—progressive supranuclear palsy. Age acceleration residuals were obtained by regressing DNA methylation age against chronological age and adjusting for neuronal proportions; the bar plots depict the mean value and standard error (y-axis); p-values for across group comparisons were calculated using the Kruskal–Wallis test (a–f). |


Please read our newsletters to find out more about our research:
Page updated 29.02.2024