Point defects in ZnO
Alexey A. Sokol, Samuel A. French, Stefan T. Bromley, C. Richard
A. Catlow
Huub J. J. van Dam and Paul
Sherwood
ZnO is widely used in catalysis,
electrical devices, optoelectronics, and pharmaceuticals, which often crucially
depend on the defect properties of this versatile material. The nature of the
intrinsic defects in ZnO, however, remains elusive, and, so far, there is no
unambiguous assignment of experimental data to particular defect species (with
the possible exception of the positron annihilation spectroscopic evidence for
Zn vacancies - see references [[1]-[3]]
and references therein). Theoretical work is therefore essential to complement
the extensive body of experimental data.
We have investigated first the
intrinsic point defects in ZnO and secondly, H, N, P, Li, Fe, Cu, Al and In
impurity centres [[4]].
Atomic and electronic structures as well as defect formation energies have been
obtained for the main oxidation states of all the defects using our embedded
cluster, hybrid Quantum Mechanical / Molecular Mechanical approach to the
treatment of localised states in ionic solids [[5],[6]].
In contrast to a large number of recent periodic density functional
calculations, we are able to employ in these studies, significantly more
accurate QM methods, for example, those based on hybrid
exchange-correlation density functionals, which allows us to approach the limit
of chemical accuracy in the energetics of defect formation, which are given in
Table 1), where we also report energies calculated using the classical
Mott-Littleton approach.
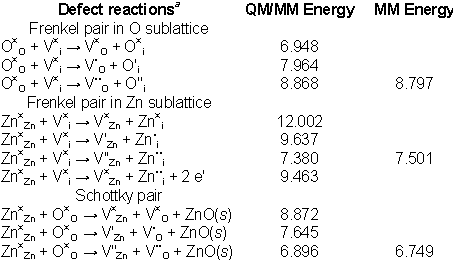
Table 1: Calculated energies for the defect pair formation
in ZnO. Pure MM energies obtained using Mott-Littleton approach and interatomic
potentials are shown for comparison for relevant charged defects.
The results show that oxygen
Frenkel pairs have low energies of formation; moreover, we note that O
interstitials have the lowest energies of formation among all intrinsic
defects, which suggests their dominance under oxidising conditions. However,
both Zn interstitials (2.2 eV) and O interstitials (1.7 eV) have
similar, relatively low energies of formation. Hence, the dominant defect
species should be decided upon by the sample history and working conditions.
Next, based on the calculated
values of the vertical ionisation potential and electron affinity, we have
determined defect levels in the band gap of the ZnO; the results are
illustrated in the figure. With these calculations we have been able to explain
the following experimentally observed phenomena:
·
We propose that the neutral and singly positively charged Zn
interstitial defect is responsible for E1
and E3 (majority) donor bands from
electric measurements.
·
Zn vacancies are proposed as the majority acceptor in
agreement with experimental assignment based on positron annihilation
spectroscopic and other studies. This defect is found to be stable in five
charge states. Exciton recombination at this defect species is proposed as a
source for main photoluminescence bands: ultraviolet (an acceptor level at 3.2 eV
in a Donor to Acceptor Photoluminescence, DAP, transition); green (a triplet
level at 2.5 eV), and red (at 1.9-2.0 eV).
·
The neutral O interstitial in a split-interstitial peroxy
configuration (at 2.8 eV) should also contribute to blue and green
luminescence by an exciton recombination and DAP transition from donor Zn
interstitials.
·
O vacancies could not be a source of green luminescence, but
could contribute to near-gap (UV) and red-orange luminescence bands (at
2.1 eV and below) via the exciton recombination mechanism.
·
Our calculations confirm that Cu, which is stable in ZnO in
two charge states, is an efficient electron scavenger. The singly negatively
charged Cu impurity is proposed as an E4 donor. Calculated defect levels (at
2.7 and 0.55 eV) are in good agreement with experiment, which established
Cu as a distinct source of green luminescence from ZnO.
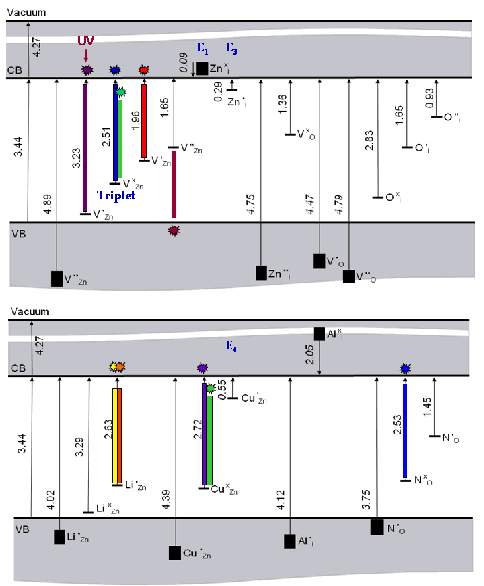
Calculated energy levels of intrinsic and extrinsic defects in ZnO (in eV)
Our calculations on impurities show their donor and acceptor properties in agreement with experiment where available. Curiously, our calculations suggest that a number of such extrinsic defects, for example, Li and N, can also contribute to the green luminescence band, the origins of which caused much controversy in the literature.
These studies are currently continuing on other extrinsic defects and defect complexes.