This is only required
by a dataset with three-body forces. This is not used at present but is
included for future development.
| Operands | none |
This instructs the
program to call the three-body bond input and geometry setup subroutine.
This has two sub-directives, ANGA
and ENDS.
| Operand types | I | 2A | 2A | 2A |
| Operand names | ILABEL | LABELI | LABELJ | LABELK |
- ILABEL
- is the label of the three-body bond type. It must be the same integer as used in the BOHA sub-directive of the potential input routine to define the three-body force constant.
- LABELI, LABELJ and LABELK
- are three species
labels (See BASI). The three-body bond
angle acts about species i (i.e. the angle is
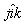 ).
).
| F | F | F | < F | F | F > |
| IJMAX | IKMAX | JKMAX | IJMIN | IKMIN | JKMIN |
where IJMAX, IKMAX
and JKMAX are the cutoffs of the three-body bond for the ij, ik and jk
distances respectively. Remember that the centre atom of the bond is i.
The cutoffs are input in Angstroms. The optional parameters IJMIN, IKMIN
and JKMIN are the minimum acceptable bond length. Their default value
is 0.0 if they are not given. The program will then find all three-body
bonds in the unit cell for which
| IJMIN | < | < | IJMAX | |
| IKMIN | < | < | IKMAX | |
| JKMIN | < | < | JKMAX |
and for which the species i, j, and k are the correct types given by LABELI, LABELJ and LABELK. i will always be one of the unit cell basis atoms, but j and k may be translated by a lattice vector to another unit cell.
Some comment is necessary about the symmetry the program assumes with respect to interchange of pairs of particles. All of the three-body potential types except TRID are symmetric with respect to interchange of one pair of atoms only, the atoms j and k. In setting up the list of three-body interactions, the program will assume that the potential is of this type even for the TRID potential. This means that for TRID potentials, too many bonds may be set up by the program. The force constants should be adjusted accordingly. The possible cases are listed below
- All three species types different. In this case there is no problem. The program will enumerate a single bond A B C.
- Species j and k the same but different from i. Again there is no problem in this case, the program will enumerate a single bond A B B.
- Species i and j are the same, but species k differs. In this case the program will enumerate A1 A2 B and A2 A1 B as separate bonds, since the angle about the centre species is different in the two cases. The cutoffs may prevent both cases being enumerated. However for the TRID potential the two cases are the same. Force constants for these cases will need to be input divided by 2 for the TRID potential.
- All three species the same. In this case the program will enumerate A1 A2 A3, A2 A1 A3 and A3 A1 A2 provided the cutoffs permit.
One and only one
ANGA directive must be given for each three-body bond type (ILABEL), up
to the same maximum of MAXTHB.
Printed output
If PRIN BOND 1 is set, the following is outputBONDS WITH 3 BODY INDEX ILABEL ARE CALCULATED BETWEEN
| LABELI | LABELJ | LABELK | |||
| CUTOFFS ARE (IN ANGSTROMS) | |||||
| RIJMAX | RIKMAX | RJKMAX | RIJMIN | RIKMIN | RJKMIN |
| RIJMAX | RIKMAX | RJKMAX | RIJMIN | RIKMIN | RJKMIN |
| LIST OF BONDS | ||||||||||||||
| INDEX | I | J | K | |||||||||||
| X | Y | Z | X | Y | Z | X | Y | Z | RIJ | RIK | RJK | THETA0 | THETA | |
Coordinates output are orthonormal coordinates. RIJ, RIK and RJK are in Å. THETA and THETA0 are output in degrees.
Error messages
ERROR - ONLY ONE THBO DIRECTIVE IS PERMITTEDOnly one THBO directive is permitted before the START PLUT directive. THBO may be repeated after the START PLUT directive. This is most useful with PRIN BOND 2, so that the relaxed bond lengths and angles are printed out after the perfect lattice relaxation.
ERROR - POTENTIALS MUST BE READ IN BEFORE 3 OR 4 BODY BONDSThe dataset card order is incorrect. Ensure that the POTE directive comes before the THBO directive.
ERROR - THREE BODY BONDS MUST BE INPUT BEFORE FOUR BODY BONDSThe dataset card order is incorrect. The directive THBO must be placed before TORS.
INVALID DIRECTIVE IN THREE BODY FORCE INPUTA sub-directive has been found which is neither ANGA nor ENDS.
TOO MANY BONDS READ INThe memory requirements exceed that available. On the CRAY, rerun the job with a larger value for the MFL= parameter on the JOB card.
INITIAL BOND ANGLE DEVIATES FROM EQUILIBRIUM BY MORE THAN PERMITTED TOLERANCEA bond angle has been found in the unit cell for which THETA - THETA0 is greater than ACCTH. (See REGION RADII) Check that the cutoffs supplied after the ANGA directive are not too large.
ERROR - A THREE BODY BOND HAS BEEN INPUT WHICH IS LINEARThis is caused by a unit cell with a linear bond. Check that the cutoffs given after the ANGA directive are not too large. This error message will only be given for those three-body potential types for which linear bonds are prohibited.